

Introduction
Materials and methods
Materials
Sample preparation
Cell culture and treatment
Cell viability assay
Measurement of NO production
Isolation of cytosol and nucleus fraction
SDS-PAGE and Western blot analysis
Reverse transcriptase-polymerase chain reaction (RT-PCR)
Transient transfection and luciferase activity
Statistical analysis
Results and Discussion
Effect of RHS-E70 on LPS-mediated NO production, and expression of iNOS and IL-1β in LPS-stimulated RAW264.7 cells
Inhibitory of RHS-E70 on NF-κB signaling activation in LPS-stimulated RAW264.7 cells
Inhibitory of RHS-E70 on MAPK/ATF2 signaling in LPS-stimulated RAW264.7 cells
Introduction
Although inflammation has been regarded as a self-protective response against a variety of harmful stimuli such as damaged cells, irritants and pathogens, abnormal inflammation induces various human diseases such as arthritis, asthma, cancer and multiple sclerosis (Guzik et al., 2003; Rankin, 2004). Nitric oxide (NO) produced by NO synthases (NOS) is known to be an important molecule involved in immune and inflammatory responses (Guzik et al., 2003). Among NOS, constitutive NOS is detected in neuronal tissues and vascular endothelial cells called as nNOS and eNOS, respectively. However, inducible NOS (iNOS) is expressed in various cell types such as macrophages under inflammatory stimulation (Nathan, 1997). Because excessive amount of NO produced by iNOS is associated with the chronic inflammatory process and related human diseases, the inhibition of iNOS and NO production has been regarded as a strategy for the treatment of inflammatory diseases (Aktan, 2004; Chen et al., 2015). In addition, interleukin-1β (IL-1β) induces excessive NO production by activating iNOS expression (Kanno et al., 1994; Muhl and Pfeilschifter, 1995). Thus, the inhibition of IL-1β-induced iNOS expression has been regarded as one of the pharmacological approaches for human inflammatory diseases (Muhl and Pfeilschifter, 1995).
Hibiscus syriacus (H. syriacus) as the national flower of Korea has been used as the herbal medicine in Asia (Hse et al., 2015). Recent studies shows that H. syriacus has antipyretic, anthelmintic, antioxidant, anticancer and antifungal properties (Kwon et al., 2003; Maganha et al., 2010; Hse et al., 2015).
In this study, we aimed to investigate the anti-inflammatory effect of 70% ethanol extracts from the root of Hibiscus syriacus, and to elucidate the potential mechanism associated with its anti-inflammatory activity.
Materials and methods
Materials
Dulbecco’s Modified Eagle medium (DMEM)/F-12 1:1 Modified medium (DMEM/F-12) for the cell culture was purchased from Lonza (Walkersville, MD, USA). Lipopolysaccharide (LPS) was purchased from Sigma Aldrich (St. Louis, MO, USA). Antibodies against IκB-α, phospho-IκB-α, p65, phospho-ERK1/2, ERK1/2, phospho-p38, p38, and β-actin were purchased from Cell Signaling (Bervely, MA, USA).
Sample preparation
The root of Hibiscus syriacus (H. syriacus) were friendly provided from Forest Medicinal Resources Research Center, National Institute of Forest Science, Yongju, Korea and formally identified by Ho Jun Son as a researcher of Forest Medicinal Resources Research Center, Korea. Twenty gram of dried root of H. syriacus was extracted with 100 ㎖ of 70% ethanol for 72 h under shaking at the room temperature. After 72 h, the ethanol-soluble fraction was filtered and concentrated to approximately 30 ㎖ volume using a vacuum evaporator and then freeze-dried. The ethanol extracts from the root of H. syriacus (RHS-E70) were kept in a refrigerator until use.
Cell culture and treatment
Mouse macrophage cell line, RAW264.7 was purchased from Korean Cell Line Bank (Seoul, Korea) and grown in DMEM/F-12 supplemented with 10% fatal bovine serum (FBS), 100 U/㎖ penicillin and 100 ㎍/㎖ streptomycin. The cells were maintained at 37℃ under a humidified atmosphere of 5% CO2. RHS-E70 was dissolved in dimethyl sulfoxide (DMSO) and treated to cells. DMSO was used as a vehicle and the final DMSO concentration did not exceed 0.1% (v/v).
Cell viability assay
Cell viability was evaluated by MTT assay. Briefly, cells were plated at a density of 3 × 104 cells/well in 96-well plate and incubated for 24 h. The cells were treated with RHS-E70 at the indicated concentrations for 24 h. Then, the cells were incubated with 50 ㎕ of MTT solution (1 ㎎/㎖) for an additional 2 h. The resulting crystals were dissolved in DMSO. The formation of formazan was measured by reading absorbance at a wavelength of 570 ㎚ using UV/Visible spectrophotometer (Human Cop., Xma-3000PC, Seoul, Korea).
Measurement of NO production
RWA264.7 cells were seeded at 1 × 105 cells/well in 96-well plate overnight. The cells were pretreated with RHS-E70 at the indicated concentrations for 6 h and then co-treated with LPS (1 ㎍/㎖) for 18 h. NO levels were measured by Griess assay. Briefly, 100 ㎕ of the cell culture supernatants were mixed with 100 ㎕ of Griess reagent (Sigma Aldrich, St. Louis, MO, USA) and followed by incubation for 15 min at the room temperature. After 15 min, absorbance values were measured using a UV/Visible spectrophotometer (Human Cop., Xma-3000PC, Seoul, Korea) at 540 ㎚.
Isolation of cytosol and nucleus fraction
Cytosol and nuclear fractions of cells were prepared using a nuclear extract kit (Active Motif, Carlsbad, CA, USA) according to the manufacturer's protocols. Briefly, RAW264.7 cells after the treatment were harvested with 1 × cold hypotonic buffer and incubated at 4℃ for 15 min. After adding detergent and vortexing for 10 s, the cells were centrifuged at 14,000 g for 1 min at 4℃ and the supernatants (cytoplasmic fraction) were collected and stored at -80℃ for further analysis. The cell pellets were used for nuclear fraction collection. Cell pellets were re-suspended with lysis buffer by pipetting up and down, and incubated at 4℃ for 30 min under shaking. After 30 min, nuclear suspensions were centrifuged at 14,000 g for 10 min at 4℃, and the supernatants (nuclear fraction) were stored at -80℃ for further analysis.
SDS-PAGE and Western blot analysis
After treatment, the cells were washed with 1×phosphate-buffered saline (PBS), and lysed in radioimmunoprecipitation assay (RIPA) buffer (Boston Bio Products, Ashland, MA, USA) supplemented with protease inhibitor cocktail (Sigma-Aldrich) and phosphatase inhibitor cocktail (Sigma-Aldrich), and centrifuged at 15,000 × rpm for 10 min at 4℃. Protein concentration was determined by the bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL, USA). The proteins were separated on SDS-PAGE and transferred to PVDF membrane (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The membranes were blocked for non-specific binding with 5% non-fat dry milk in Tris-buffered saline containing 0.05% Tween 20 (TBS-T) for 1h at room temperature and then incubated with specific primary antibodies in 5% non-fat dry milk at 4°C overnight. After three washes with TBS-T, the membranes were incubated with horse radish peroxidase (HRP)-conjugated immunoglobulin G (IgG) for 1 h at room temperature and chemiluminescence was detected with ECL Western blotting substrate (Amersham Biosciences, Piscataway, NJ, USA) and visualized in Polaroid film.
Reverse transcriptase-polymerase chain reaction (RT-PCR)
After treatment, total RNA was prepared using a RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and total RNA (1 ㎍) was reverse-transcribed using a Verso cDNA Kit (Thermo Scientific, Pittsburgh, PA, USA) according to the manufacturer's protocol for cDNA synthesis. PCR was carried out using PCR Master Mix Kit (Promega, Madison, WI, USA) with mouse primers for iNOS, IL-1β and GAPDH as followed : mouse iNOS: forward 5’-ttgtgcatcgacctaggctggaa-3’ and reverse 5’-gacctttcgcattagcatggaagc-3’, mouse IL-1β: forward 5’-ggcaggcagtatcactcatt-3’ and reverse 5’-cccaaggccacaggtattt-3’, GAPDH: forward 5’-ggactgtggtcatgagcccttcca-3’ and reverse 5’-actcacggcaaattcaacggcac-3’. The PCR results were visualized using agarose gel electrophoresis.
Transient transfection and luciferase activity
Transient transfection for luciferase activity was carried out using the PolyJet DNA transfection reagent (SignaGen Laboratories, Ijamsville, MD, USA) according to the manufacturers' instruction. Cells were seeded in 12-well plates at a concentration of 2 × 105 cells/well. After growth overnight, plasmid mixtures containing 1 ㎍ of the NF-κB luciferase constructs (Addgene, Cambridge, MA, USA) and 0.1 ㎍ of pRL-null vector were transfected for 24 h. After the treatment, the cells were then harvested in 1 × luciferase lysis buffer, and luciferase activity was normalized to the pRL-null luciferase activity using a dual-luciferase assay kit (Promega, Madison, WI, USA).
Statistical analysis
All the data are shown as mean ± SD (standard deviation). Statistical analysis was performed with one-way ANOVA followed by Dunnett's test. Differences with *P or #P < 0.05 were considered statistically significant.
Results and Discussion
Effect of RHS-E70 on LPS-mediated NO production, and expression of iNOS and IL-1β in LPS-stimulated RAW264.7 cells
Because excessive NO production has been reported to be related to chronic inflammation and related human diseases (Aktan, 2004; Chen et al., 2015), the inhibitory effect of RHS-E70 on NO production was determined to evaluate the anti-inflammatory activity of RHS-E70. As shown in Fig. 1A, excessive NO production was observed in the cell treated with LPS alone. However, RHS-E70 dose-dependently blocked LPS-mediated NO production in RAW264.7 cells. To exclude that inhibition of NO production by RHS-E70 results from the reduction of the cell viability, we evaluated the effect of RHS-E70 on the viability of RAW264.7 cells. As shown in Fig. 1B, RHS-E70 increased the cell viability by 108.9 ± 1.3% at 50 ㎍/㎖ and 113.6 ± 1.31.9% at 100 ㎍/㎖, respectively.
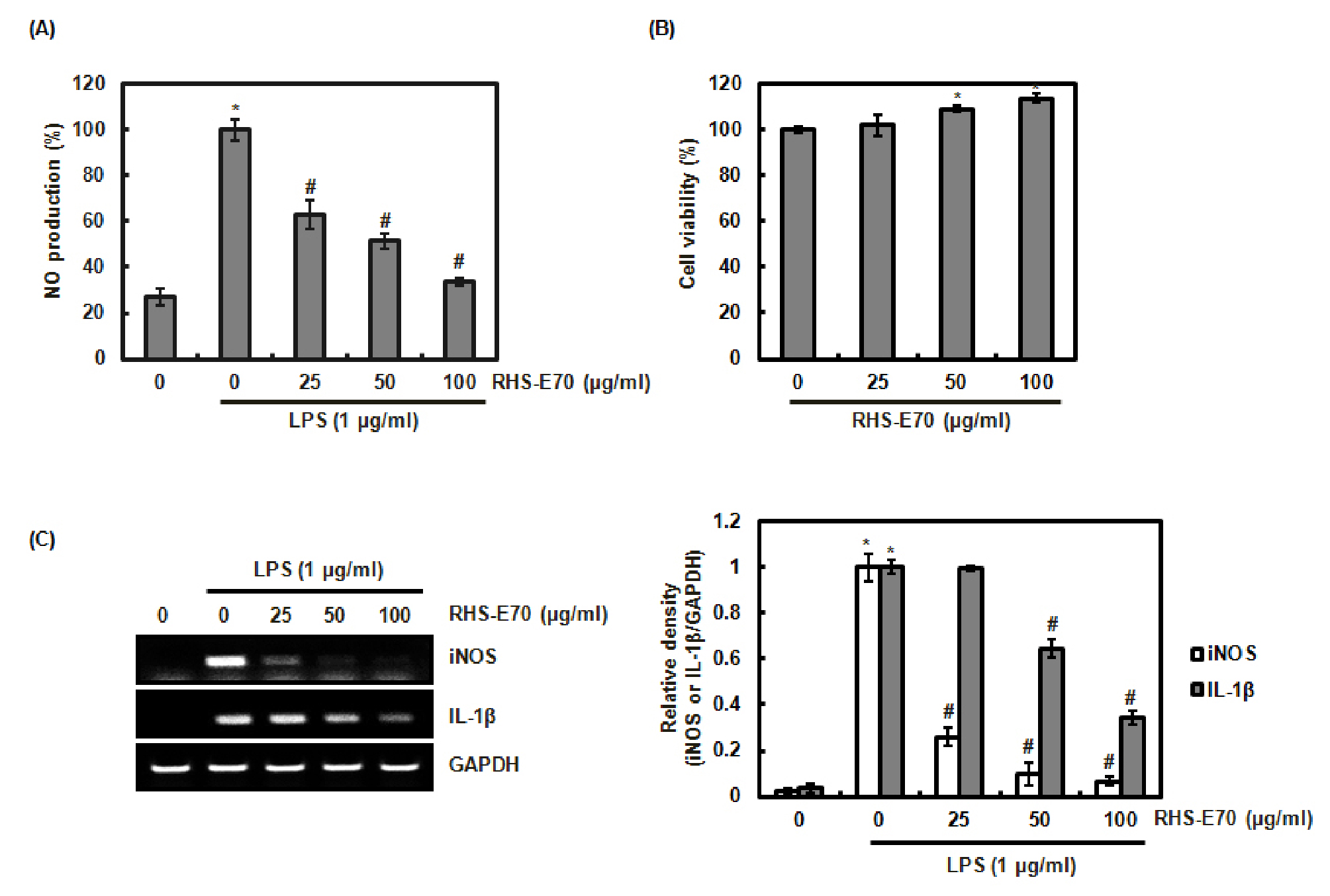
Fig. 1.
Inhibitory effect of RHS-E70 on NO production, and the expressionof iNOS and IL-1β in LPS-stimulated RAW264.7 cells. (A) RAW264.7 cells were pretreated with RHS-E70 for 6 h and then co-treated with LPS (1 ㎍/㎖) for 18 h. The determination of NO production was measured by Griess assay. (B) RAW264.7 cells were treated with RHS-E70 for 24 h. the cell viability was measured by MTT assay. (C) RAW264.7 cells were pretreated with RHS-E70 for 6 h and then co-treated with LPS (1 ㎍/㎖) for 18 h. Total RNA was prepared. GAPDH was used as internal control for RT-PCR. *P < 0.05 compared to the cells without the treatment, and #P < 0.05 compared to the cells treated with LPS alone.
Because iNOS and IL-1β result in the excessive production of NO (Muhl and Pfeilschifter, 1995; Aktan, 2004; Chen et al., 2015), we investigated the effect of RHS-E70 on the expression of iNOS and IL-1β to determine whether the inhibition of NO production by iNOS and IL-1β results from the regulation of iNOS and IL-1β expression. As a result, the overexpression of iNOS and IL-1β was observed in the cell treated with LPS alone, while RHS-E70 dose-dependently suppressed iNOS and IL-1β expression in LPS-stimulated RAW264.7 cells (Fig. 1C). These result indicate that RHS-E70 may block excessive NO production by inhibiting NOS and IL-1β expression, which shows that RHS-E70 may exert anti-inflammatory effect.
Inhibitory of RHS-E70 on NF-κB signaling activation in LPS-stimulated RAW264.7 cells
For searching the potential anti-inflammatory agents, inhibition of the inflammatory mediators has been regarded to be important, but it is also required to elucidate the mechanisms that regulate the expression of inflammatory mediators. As a well-known transcription factor involved in the inflammatory response, nuclear factor-κB (NF-κB) upregulates a variety of inflammatory mediators such as iNOS and IL-1β (Chen et al., 2003; Gloire et al., 2006; Olivera et al., 2012). NF-κB signaling is regulated by the degradation of IκB-α through its phosphorylation and subsequent ubiquitination (Baeuerle and Baltimore, 1996; Olivera et al., 2012; Gantke et al., 2012). Under the degradation of IκB-α, NF-κB translocates to the nucleus and binds to inflammatory mediator-related genes, which results in the expression of inflammatory mediators (Baeuerle and Baltimore, 1996; Olivera et al., 2012; Gantke et al., 2012). Thus, NF-κB signaling have been viewed as the potential target for the development of anti-inflammatory agents.
To determine whether RHS-E70 inhibits NF-κB signaling, we firstly investigated the effect of RHS-E70 on LPS-mediated phosphorylation and degradation of IκB-α. As shown in Fig. 2A and 2B, LPS induced IκB-α phosphorylation and subsequent degradation of IκB-α. However, RHS-E70 dose-dependently inhibited the phosphorylation and degradation of IκB-α. Next, we investigated whether the inhibition of IκB-α degradation by RHS-E70 contributes to the attenuation of p65 nuclear translocation. As a result, decreased level of the nuclear p65 was observed in the cells treated with RHS-E70 (Fig. 2C), which resulted in the inhibition of NF-κB activation (Fig. 2D).
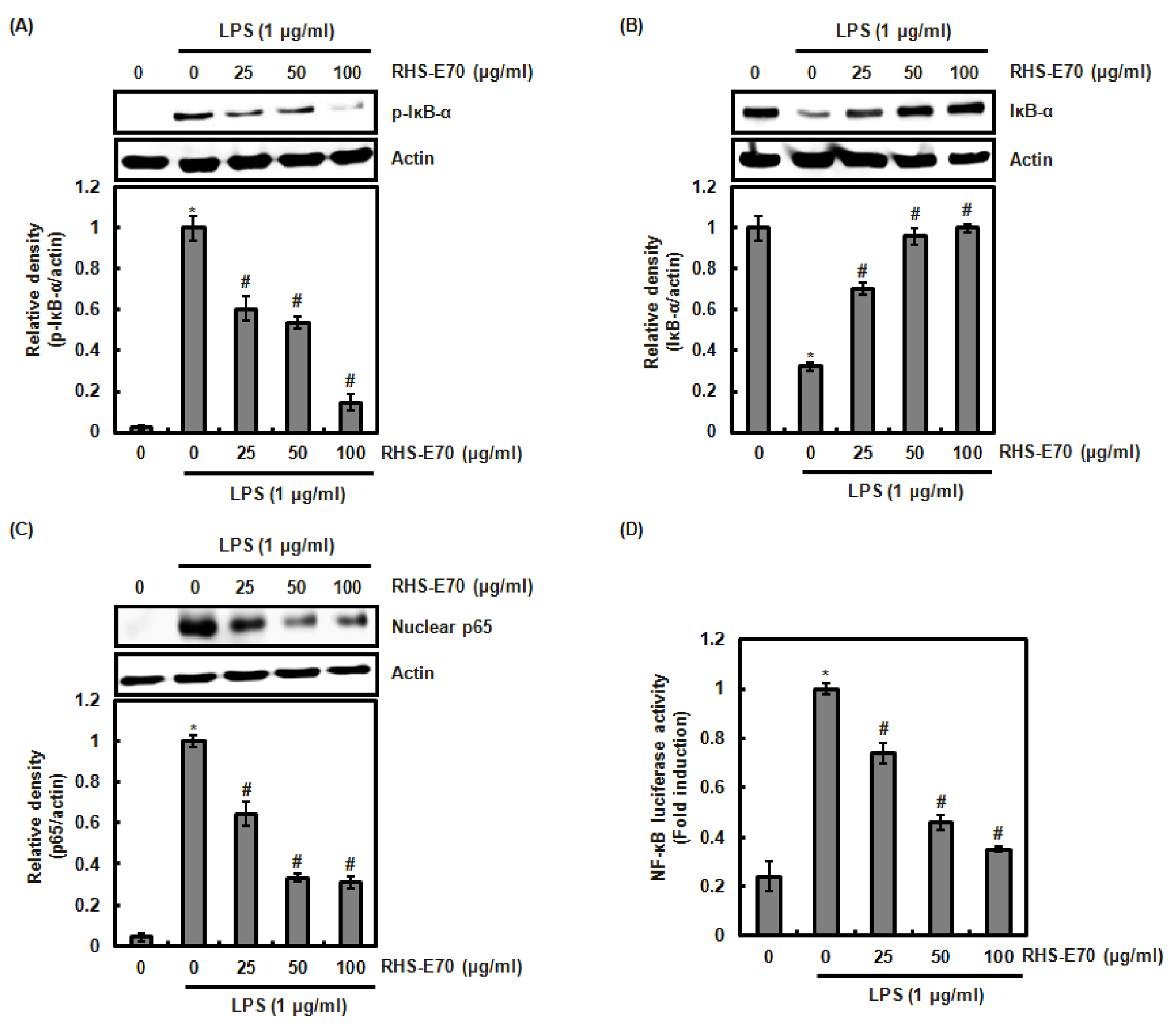
Fig. 2.
Inhibitory effect of RHS-E70 on NF-κB signaling in LPS-stimulated RAW264.7 cells. (A) RAW264.7 cells were pretreated with RHS-E70 for 6 h and then co-treated with LPS (1 ㎍/㎖) for 5 min. (B) RAW264.7 cells were pretreated with RHS-E70 for 6 h and then co-treated with LPS (1 ㎍/㎖) for 20 min. (C) RAW264.7 cells were pretreated with RHS-E70 for 6 h and then co-treated with LPS (1 ㎍/㎖) for 30 min. After the treatment, the cytosol and nucleus were prepared. For Western blot analysis, the cell lysates were subjected to SDS-PAGE and the Western blot was performed using antibodies against p-IκB-α, IκB-α and p65. Actin was used as internal control. (D) RAW264.7 cells were co-transfected with NF-κB luciferase constructs and pRL-null. The cells were pretreated with RHS-E70 for 6 h and then co-treated with LPS (1 ㎍/㎖) for 18 h. Luciferase activity for NF-κB was measured as a ratio of firefly luciferase signal/renilla luciferase signal using a dual luciferase assay kit. *P < 0.05 compared to the cells without the treatment, and #P < 0.05 compared to the cells treated with LPS alone.
These result indicate that the inhibition of NF-κB signaling by RHS-E70 may contribute to the attenuation of the inflammatory mediators such as iNOS and IL-1β.
Inhibitory of RHS-E70 on MAPK/ATF2 signaling in LPS-stimulated RAW264.7 cells
Besides NF-κB signaling, The mitogen-activated protein kinases (MAPK) also are involved in the production of the inflammatory mediators (Kaminska, 2005). In addition, MAPK have been reported to activate NF-κB pathway (Surh et al., 2001). The inflammatory stimuli induce MAPK activation by phosphorylating ERK1/2, p38 and JNK, which results in the expression of the inflammatory mediators such as iNOS and IL-1β (Nakagawa and Maeda, 2012). Thus, MAPK has been regarded as the potential target for the development of anti-inflammatory agents. Thus, we investigated whether RHS-E70 inhibits MAPK activation. As shown in Fig. 3A, RHS-E70 attenuated LPS-induced phosphorylation of ERK1/2 and p38 compared to LPS treatment alone, which indicates that RHS-E70 may suppress MAPK activation.
There is growing evidence that the phosphorylation and subsequent nuclear accumulation of ativating transcription factor 2 (ATF2) is required to induce the expression of the inflammatory mediators by MAPK (Yu et al., 2014). Because RHS-E70 suppressed MAPK activation, we investigated whether the phosphorylation and nuclear accumulation of ATF2 is inhibited by RHS-E70. As shown in Fig. 3B and 3C, RHS-E70 dose-dependently attenuated ATF2 phosphorylation and nuclear accumulation. These data indicate that the inhibition of MAPK activation by RHS-E70 may contribute to the mitigation of ATF2 nuclear accumulation, which exerts anti-inflammatory effect of RHS-E70.
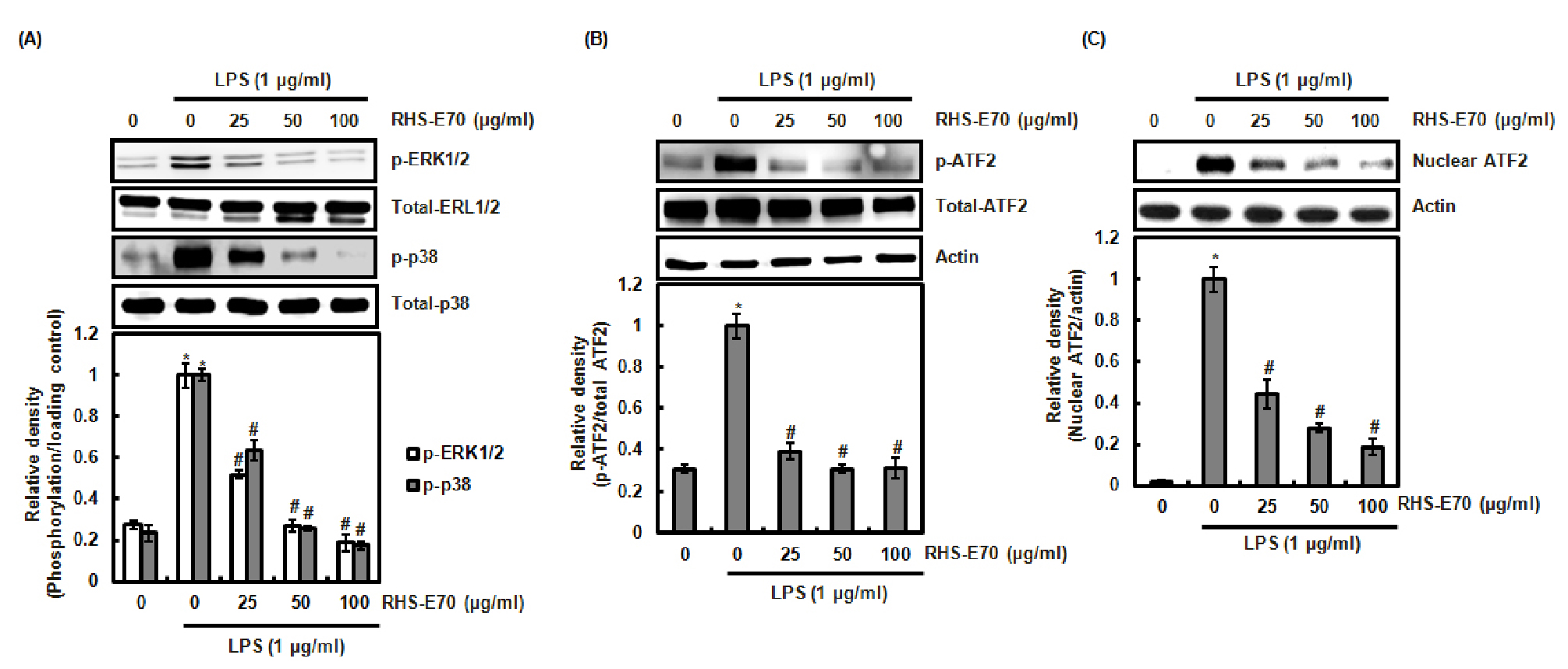
Fig. 3.
Inhibitory effect of RHS-E70 on MAPK/ATF2 signaling. (A, B) RAW264.7 cells were pretreated with RHS-E70 for 6 h and then co-treated with LPS (1 ㎍/㎖) for 20 min. (C) RAW264.7 cells were pretreated with RHS-E70 for 6 h and then co-treated with LPS (1 ㎍/㎖) for 40 min. After the treatment, the cytosol and nucleus were prepared. For Western blot analysis, the cell lysates were subjected to SDS-PAGE and the Western blot was performed using antibodies against p-ERK1/2, ERK1/2, p-p38, p38, p-ATF2 and ATF2 Actin, ERK1/2, p38 or ATF2 was used as internal control. *P < 0.05 compared to the cells without the treatment, and #P < 0.05 compared to the cells treated with LPS alone.
Taken together, these results demonstrated that RHS-E70 blocked NO production via inhibiting iNOS and IL-1β expression through suppressing the activation of NF-κB and MAPK/ATF2. The anti-inflammatory effect and potential mechanisms of RHS-E70 will provide the potential usage of RHS-E70 in the anti-inflammatory drug development.
